

新版MDR推迟一年实施!欧盟官方防护装备合规指南来了
欧盟通过议案
新版医疗器械法规将推迟一年实施
4月3日,欧盟通过议案,新版医疗器械法规(MDR)将推迟一年实施,出口企业可暂不考虑医疗器械新旧CE证书换版问题。

3月13日欧盟出台指令(EU) 2020/403,指令中提到:在疫情期间,允许部分防疫物资(如一类灭菌的医用口罩)在符合安全有效的情况下,即使尚未获得CE认证,也可以在欧盟市场上市销售,成员国主管当局也可在疫情期间评估和组织采购没有CE标记的医疗器械,但出口企业应注意到,该指令有附加条件,即上述未获得CE的产品仅可提供给医疗工作者使用,不能在市场上流通销售。同时市场将会重点抽查防疫相关医疗器械,以防止不合格产品引起严重风险。
为降低新冠肺炎影响,增加防护装备生产和供应,3月30日欧盟委员会发布《防护装备合格评定程序》指南,采用问答形式详细说明了制造商将防护产品投放到欧盟市场所需适用的欧盟法律框架和步骤,为新加入和有意转产防护设备的企业生产销售提供了合规指南。指南做出的官方说明对解决我国近期防护装备生产和出口企业高度关切质量保障、合规认证问题具有参考意义。主要内容如下: QUESTION1 问题:防护装备适用哪些欧盟法律框架? 回答:非医用个人防护装备,如FFP类型口罩等适用于《个人防护装备(PPE)法规》,即EU 2016/425(以下简称PPE)。医用防护装备,例如医用手套、外科口罩、重症监护和其他医疗设备等,则适用医疗器械法规93/42/EEC(以下简称MDD)指令范围内。该指令将于2020年5月26日被新的欧盟医疗器械法规 EU 2017/745(以下简称MDR)取代。但是,3月25日,欧盟委员会宣布一项推迟1年实施MDR法规的提案,并在4月初提交,以便欧盟议会和理事会5月底前通过。这两个法规体系都协调统一了其涵盖产品的性能要求,以确保用户的健康和安全。因此,根据这些法规制造的产品可以在整个欧盟市场自由流通,欧盟成员国不得对此类产品的制造和投放市场提出额外的和不同的要求。 QUESTION2 问题:生产防护装备,是否有强制性的欧盟标准? 回答:PPE和MDD法规都对其所涵盖产品的卫生、安全和性能提出了基本要求。但是,这两个欧盟法规在技术上都是中立的,没有为产品设计规定任何特定的强制性技术解决方案。因此,制造商可以使用各种技术解决方案来满足这些基本要求。 同时,PPE和MDD均为制造商提供了特定技术解决方案。这些技术解决方案在协调标准或其部分中都有详细说明,这些协调标准的采用要求已发布在欧盟公报上。如果制造商选择采用这些技术解决方案,则该产品推定符合适用的基本卫生、安全和性能要求。 QUESTION3 问题:如果制造商选择遵循EN14683标准生产外科口罩,那么谁应该执行其中规定的测试? 回答:测试是标准中规定的,可以由制造商或者由实验室代表制造商进行。从严格意义上讲,这些测试并不是投放市场之前必须执行的步骤。但是,如果制造商声称产品符合该标准,那么市场监督机构可以根据监督管理需要,按照EN14683标准规定的测试方法对样品进行测试。 QUESTION4 问题:如果制造商想要遵循EN149或EN14683标准进行生产,那么可以从哪里获得这些标准? 回答:欧洲标准的所有权归属其制定者欧洲标准化组织。通常,制造商必须从欧洲标准化组织的国家成员(即国家标准化机构)购买所需的标准。 但是,为确保欧洲工业能够快速应对由于新冠肺炎COVID-19暴发带来的防护装备增长需求,欧盟委员会已经与欧洲标准化组织CEN达成协议,将14个标准(包括EN149和EN14683)由国家标准化机构免费和完整地提供给制造商。 制造商可以从国家标准化机构的在线目录中免费下载EN149和EN14683标准。完整列表及其相应网站的链接如下:https://standards.cen.eu/dyn/www/f?p=CENWEB:5. QUESTION5 问题:防护装备是否可以遵循其他标准? 回答:只要能够符合产品适用欧盟法律框架中规定的基本要求,就可以使用任何标准或特定的技术解决方案。尽管协调标准(例如,口罩的标准EN149和EN14683)往往是欧盟工业使用最广泛的技术解决方案,但还有其他一些特定的技术解决方案可以确保同等水平的安全性。《世界卫生组织防护装备选择指南》在这方面提供了指引。 但是,与使用协调标准(EN149和EN14683)不同的是,如果产品在PPE法规管辖范围内,制造商选择遵循世界卫生组织WHO提供的替代标准,则应由公告机构(第三方测试机构)对产品样品进行测试。 为了应对新冠肺炎COVID-19,欧盟委员会于2020年3月13日发布了《新冠肺炎COVID-19威胁下合格评定程序和市场监督管理的建议》(以下简称《建议》),以促进新产品在欧盟市场上的快速普及。一方面,欧盟委员会敦促所有公告机构(第三方测试机构)对制造商提交的与新冠肺炎COVID-19相关产品的新申请进行优先处理,向公告机构强调世界卫生组织的指南可以作为替代性的适用技术解决方案。 另一方面,《建议》提供了两种情况,即使合格评定程序尚未最终确定,或者在某些特殊情况下合评定程序刚刚启动,产品也可以投放市场: (1)如果国家市场监督机构认为欧盟市场中的防护装备能够遵循欧盟法律中规定的基本要求以确保足够高的卫生和安全水平,即使合格评定程序(包括CE标记的粘贴)尚未完全完成,监督管理机构也应允许这些产品进入欧盟市场。 (2)在特殊情况下,如果满足下述全部要求,即使尚未启动认证程序并且没有在产品上贴有CE标志,也可以将产品投放市场: (a)产品是按照EN标准之一或是世界卫生组织WHO指南中提及的任何其他标准,或是能够确保足够高的安全水平的技术解决方案制造的; (b)产品是由相关成员国当局组织采购的一部分; (c)产品仅提供给健康护理工作者; (d)产品仅在当前的新冠肺炎期间提供; (e)产品未进入常规分销渠道,不会提供给其他用户。 根据上述第(a)点的要求,对特定技术解决方案的评估应由市场监督机构在成员国有关当局组织的购买过程中执行。 QUESTION6 问题:在产品投放市场之前,有哪些授权或是强制性认证需要完成? 回答:外科/医用口罩,检查手套和防护服(以非灭菌状态提供)是“ I类医疗设备”。因此,在投放市场前它们不需要公告机构(第三方测试机构)的强制性认证。但制造商必须证明产品符合其适用的要求。这种制度本质上是一种自我认证。如果以无菌状态提供此类设备,则需要将其归为较高风险类别,并且需要由公告机构进行合格评定。 相反,应对新冠肺炎COVID-19疫情而使用的PPE法规范围内口罩和其他用品,则属于“ III类PPE”产品。因此,在所有情况下都必须由公告机构(第三方测试机构)认证。在将产品投放市场之前,需要将其样品提供给此类机构进行评估。但是,根据PPE法规,一旦对初始样品进行了测试,就没有必要对从生产线出来的每个产品都进行相同的测试,而是执行产品的随机抽查或其他类似的生产控制程序。 QUESTION7 问题:产品是否在所有情况下都应该贴上CE标志? 回答:粘贴CE标志是产品投放市场前的最后一步,标志着所有程序已经完成。对于某些医疗设备,制造商可以粘贴该标记,而不必涉及第三方测试机构( 如,I类医疗设备)。对于个人防护装备,通常应在公告机构(第三方测试机构)对产品的第一批样品进行评估和批准后,由制造商粘贴CE标记(如,III类个人防护物资)。但是,在新冠肺炎COVID-19这种特定情况下(如,《建议》提及的两种情况),这些要求可能会有所降低。值得注意的是,根据PPE和MDD/MDR法规,每个产品都应贴上CE标志。
QUESTION8
问题:是否有关于制造口罩的确切材料的规格?
回答:欧盟法律框架或协调标准EN149和EN 14683均未对所使用的材料强加任何强制性规范。法律框架实质上引入了性能要求,相关标准对此进行了详细说明。因此,由制造商自己决定选择何种材料。
实际生产中,对于FFP型口罩较多使用以下材料:
(1)过滤层:聚丙烯;
(2)阀门(如果适用):橡胶或聚丙烯;
(3)带子:聚酯纤维,聚异戊二烯,莱卡;
(4)密封:聚乙烯泡沫,聚氨酯等;
(5)夹子/U型钉(如果适用):铝,钢。
对于外科口罩,建议组合使用熔喷和纺粘织物的非织造材料(S-M-S类型)。织物通常是聚丙烯。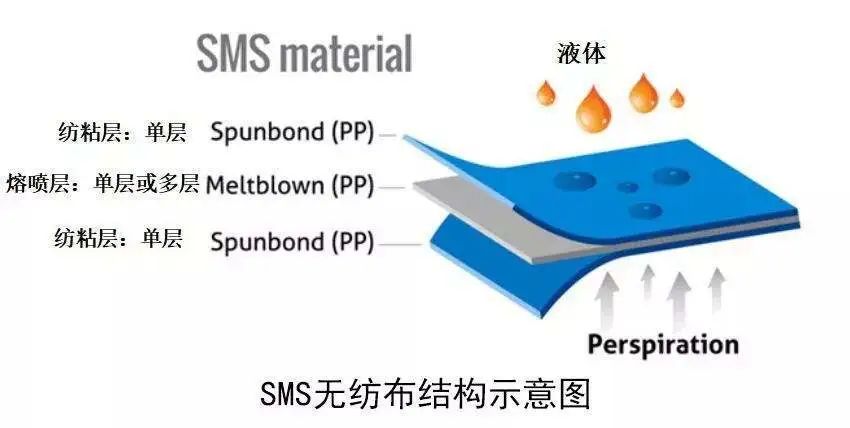
联系我们
CONTACT US
联系我们CONTACT US
| 业务常用联系方式 公司固定联系方式 | 


|



